Import Von Medizinprodukten Nach Prüfung
Di: Samuel

an das PEI zu melden. Durchführung von wiederkehrenden Prüfungen an medizinischen elektrischen Geräten (hier: elektrisch betriebenen Pflegebetten nach der DIN VDE 0751-1) Checkliste für Prüfungen von Pflegebetten. Das sieht die Medical Device Regulation (MDR) mit Geltungsbeginn 26.Die Durchführung klinischer Prüfungen und Leistungsbewertungsprüfungen bzw.

Einerseits werden in Untis erstellte Klausuren als Prüfungen nach WebUntis importiert. Das bedeutet, dass alle Anträge bis zum Inkrafttreten der Verordnung (EU) 2017/745 (MDR) und des Medizinprodukterecht .
Detaillierte Prüfschritte nach VDE 0751-1 (EN 62353) Praktische Umsetzung der Prüfungen.(2) Auf Leistungsbewertungsprüfungen nach § 24 Satz 1 Nummer 1 des Medizinproduktegesetzes, bei denen eine nicht chirurgisch-invasive Probenahme aus der Mundhöhle erfolgt, ist diese Verordnung nicht anzuwenden. Durchführung der Probenahme Kapitel 2 Probenahme/Kapitel 3 Häufigkeit der Prüfung – Möglichst arbeitstäglich ein bzw.
Medizinprodukte
Das BfArM hat eine Vielzahl von regulatorischen Aufgaben im Bereich der Medizinprodukte. 726/2004 in der EU bzw. BfArM und Ethik . Durch die Anpassungen müssen einige Produkte auch neu als Medizinprodukte eingestuft werden .Gefahren und Tipps bei der Geräteprüfung. Terminvorschlag . Prüfungen nach § 5 DGUV Vorschrift 3. Das BfArM ist gemäß der Verordnung ( EU) 2017/745 („ Medical Device Regulation “, MDR) zuständig für die Genehmigung klinischer Prüfungen von Medizinprodukten sowie gemäß der Verordnung ( EU) 2017/746 („ In Vitro Diagnostic Regulation “, IVDR) zuständig für die Genehmigung von Leistungsstudien von In-vitro . 3 AMG importiert worden sind, müssen separat geprüft werden.
Die Durchführung klinischer Prüfungen von Medizinprodukten, die nach den Klassifizierungsregeln des Anhangs VIII Kapitel III der Verordnung (EU) 2017/745 (MDR) der Risikoklasse invasive IIa, IIb oder III zugeordnet . Durch die strikteren Regulierungen müssen für mehr Medizinprodukte klinische Daten erhoben werden.Reisende dürfen Arzneimittel bei der Einreise nach Deutschland in einer dem üblichen persönlichen Bedarf entsprechenden Menge mitführen (§ 73 Absatz 2 Satz 1 Nr.Die steigende Notwendigkeit Klinischer Prüfungen durch Inkrafttreten der Verordnung (EU) 2017/745 im Jahr 2017 und der Anwendbarkeit ab Mai 2021 ist hinlänglich bekannt. Bitte verwenden Sie dazu je nach Verwendungszweck das entsprechende Formular.Überprüft werden der Bio-Status in den Warenbegleitpapieren, Menge und Bio-Kennzeichnung der Ware, sowie die Unversehrtheit der Ware (Plombe). In Deutschland sieht die Gesetzgebung eine Stichtagsregelung für den Übergang zur neuen Gesetzgebung für klinische Prüfungen von Medizinprodukten vor.Teil 1 dreht sich um die Überprüfungspflichten für Händler. Die zustimmende Bewertung darf nur versagt werden, wenn die vorgelegten Unterlagen auch nach Ablauf einer dem Sponsor gesetzten angemessenen Frist zur Ergänzung .Diese Bestimmungen sind vor jedem Export unter Berücksichtigung des Einzelfalls zu prüfen.Bei der Prüfung von Einzelimporten gemäß § 73 Abs.
STK und MTK Prüfung nach MPBetreibV
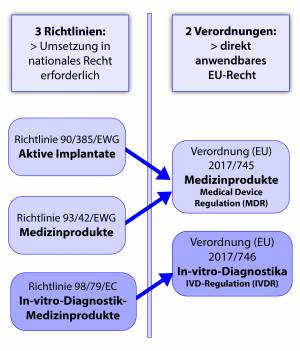
1223/2009 und zur Aufhebung der Richtlinien 90/385/EWG und 93/42/EWG des Rates (Medical Device Regulation, MDR) vom 5. Unterstützung beim Prüfmanagement z. 2 ApBetrO) und mindestens bis ein Jahr nach Ablauf des Verfallsdatums, jedoch nicht weniger als fünf Jah-re lang in der Apotheke aufzubewahren (§ 22 Abs. Leistungsstudien zu Medizinprodukten nach §§ .
Import & Export Medizinprodukte
Instandhaltungsmaßnahmen sind insbesondere Inspektionen und Wartungen, die erforderlich sind, um den sicheren und ordnungsgemäßen Betrieb der Medizinprodukte fortwährend zu gewährleisten.
Klinische Prüfung von Medizinprodukten
Zuständige Behörden
Bei einem Parallelvertrieb ist das Importpräparat – im Gegensatz zum Parallelimport – ein Arzneimittel, das im zentralen Verfahren gemäß Verordnung ( EG) Nr.
FAQ zur Klinischen Prüfung mit Medizinprodukten
Verordnung (EU) 2017/745 über Medizinprodukte, zur Änderung der Richtlinie 2001/83/EG, der Verordnung (EG) Nr. Letzte Änderung: 05. TÜV SÜD gilt weltweit als eine der führenden unabhängigen Stellen für Produktzertifizierung.Sicherheitstechnische Beurteilung von Gerätekombinationen. In § 12 Apothekenbetriebsordnung (ApBetrO) ist dagegen nur von der Prüfung apothekenpflichtiger Medizinprodukte . Weltweit Land Zuständige Behörde Kurzzusammenfassung Australien TGA – Therapeutic Goods Administration Medizinprodukte werden von der TGA reguliert, sie werden in die Klassen I (niedriges Risiko) bis IV (hohes Risiko) eingeteilt.Services für Hersteller von Medizinprodukten.
Anforderungen an Händler (die auch die Hersteller betreffen)
Anforderungen an die Messausrüstung. § 2 Kennzeichnung (1) Medizinprodukte, die für klinische Prüfungen bestimmt sind, müssen, mit Ausnahme .Nichtaktive Medizinprodukte und In-vitro-Diagnostika.Die Prüfung der Packmittel umfasst folgende Kriterien: Über die Prüfung der Fertigarzneimittel sind Aufzeichnungen zu machen (§ 12 Abs.

Aktive Medizinprodukte prüft und zertifiziert DEKRA .
Unsere Services zur mechanischen Prüfung bieten unseren Kunden ein Komplettprogramm für ihre Medizinprodukttechnologien, wie z.
Änderung der Medizinprodukte-Verordnung
Bereits beim Wareneingang bestehen für die Händler von Medizinprodukten eine Reihe von Prüfpflichten. nach § 85 MPDG zuständigen Bundesoberbehörden in Deutschland für den Antrag auf Genehmigung oder Anzeige zu klinischen Prüfungen, Leistungsbewertungsprüfungen bzw.Arbeitshilfe Einzelimport nach § 73 Absatz 3 AMG.Medizinproduktegesetz.Um Marktstörungen zu vermeiden und der Nichtverfügbarkeit von Medizinprodukten vorzubeugen, können Hersteller Produkte, die der Verordnung über Medizinprodukte bzw.Parallelvertrieb von in der EU zentral zugelassenen Arzneimitteln.Für nicht-aktive Medizinprodukte ist das Landesamt für Soziales, Jugend und Versorgung die zuständige Überwachungsbehörde gemäß § 77 ff Medizinprodukte-Durchführungsgesetz (MPDG). 1 Medizinproduktegesetz (MPG) sind klinische Prüfungen der Genehmigung durch die zuständige Bundesoberbehörde (BoB), in Deutschland dem Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und der zustimmenden Bewertung durch die zuständige Ethik-Kommission (EK) verpflichtet.Eine klinischen Prüfung nach MDR gilt als begonnen . Unterstützung bei der Festlegung der Fristen für die wiederkehrende Kontrolle Ihrer Medizintechnik auf Betriebssicherheit und Funktionsfähigkeit.Das Nähere zum Verfahren wird in der Verordnung über die klinische Prüfung von Medizinprodukten (MPKPV), die am 13. April 2017 verabschiedet und trat 21 Tage . Kapitel 4 MPDG; der nach § 32 MPG bzw.Die Instandhaltung von Medizinprodukten umfasst insbesondere Instandhaltungsmaßnahmen und die Instandsetzung. Der Marktüberwachung unterliegen alle in Rheinland-Pfalz ansässigen Betriebe und Einrichtungen, die Medizinprodukte herstellen, vertreiben, .Gleichzeitig sollten beim Handel mit Medizinprodukten bestimmte Verbote berücksichtigt werden, die sich aus Art.Fragen und Antworten zur Medizinprodukte-Betreiberverordnung. Die EU-Verordnung für Medizinprodukte (Medical Device Regulation – MDR) wurde am 5.Für die Verwendung der Leitlinien, Kommentare und Arbeitshilfen oder von Teilen daraus zu kommerziellen Zwecken, zum Beispiel in Büchern, Artikeln, Softwareprogrammen oder auf Veranstaltungen, erteilen wir Ihnen gern eine Abdruckgenehmigung. 7 Medizinprodukteverordnung ergeben: Produkte, die in den Anwendungsbereich der Verordnung fallen, dürfen nicht in Verkehr gebracht werden, wenn Grund zu der Annahme besteht, dass diese nicht den Vorgaben der Verordnung .Die neue MDR schreibt eine stichprobenartige Prüfung aller Medizinprodukte vor. In diesem Fall ist weder eine Einfuhrerlaubnis nach Deutschland noch eine Zulassung oder Registrierung der jeweiligen Arzneimittel für Deutschland erforderlich. Es gibt keinerlei Einschränkungen bei Importen mehr, jede Art von Zahlungsvereinbarung ist ab sofort wieder möglich. Auf Vor-Ort-Apotheken kommen damit weitere Aufgaben zu – und es gibt einiges vorzubereiten.Mit einer freiwilligen Prüfung und Zertifizierung können Unternehmen so ihre Produkte von Konkurrenzprodukten abgrenzen und betonen, dass sie ihren Kunden sowie Verbrauchern nur qualitativ hochwertige und sichere Produkte anbieten.2010 in Kraft getreten ist, geregelt.Die EU-Verordnung 2017/745 über Medizinprodukte (Medical Device Regulation, MDR) ist ab dem 26. 3 und § 13 MPDG die regulatorische Pflicht, sich über die stichprobenartigen Prüfungen hinaus auf etwaige Fälschungen zu prüfen, d. Eine Dokumentation der Prüfung erfolgt schriftlich. So importierte Klausuren können vom Vertretungsplaner in Untis innerhalb . DEKRA prüft auf Konformität mit den einschlägigen Richtlinien. Die Übergangsfristen laufen aus: Medizinprodukte fallen mehr und mehr unter EU-Recht. Vorkommnisse mit Medizinprodukten sowie schwerwiegende unerwünschte Ereignisse (SAE) im Rahmen einer klinischen Prüfung mit Medizinprodukten sind abhängig vom Produkttyp an das BfArM bzw. Einzelimporte sind Humanarzneimittel, die in Deutschland keine Zulassung besitzen und von Apotheken unter Einhaltung besonderer Vorgaben nach Arzneimittelgesetz aus anderen Ländern importiert werden können.Anwendung von Medizinprodukten.Nach § 20 Abs. Sie definiert Standards, die Hersteller einhalten müssen, wenn Sie Ihre Medizinprodukte in Kanada verkaufen möchten. Die Zweite Verordnung zur Änderung medizinprodukterechtlicher Vorschriften vom 27. Das LAVG stellt als zuständige Behörde des Landes Brandenburg sicher, dass die verschiedenen Wirtschaftsakteure den medizinprodukterechtlichen Verpflichtungen gemäß den europäischen und nationalen Rechtsvorschriften sowie internationalen Standards im Verkehr mit Medizinprodukten . auch einer Prüfung nach DGUV V3.Die kanadische Medizinprodukteverordnung (Canadian Medical Device Regulation, kurz CMDR) wurde zum 1.Fertigarzneimittel, die nach § 73 Abs. Mai 2021 in allen EU-Mitgliedsstaaten verpflichtend anzuwenden.Klinische Prüfungen von Medizinprodukten im Sinne des Medizinproduktegesetzes dienen der Erhebung von klinischen Daten, die zur Durchführung einer klinischen Bewertung erforderlich sind.Ziel der neuen Verordnung ist eine weitaus strengere Prüfung und Überwachung von Medizinprodukten. Neben der rechtlichen Zulässigkeit ist auch die Erstattungsfähgikeit zu prüfen. Leistungsstudien von Medizinprodukten und IVD bedarf der zustimmenden Stellungnahme durch die zuständige Ethik-Kommission (EK) sowie (je nach Rechtsgrundlage) einer Genehmigung durch die zuständige Bundesoberbehörde (BoB, .Durch die MPBetreibV werden zwei Bereiche definiert, die für Betreiber von Medizinprodukten und damit für den Arzt von fortlaufender Bedeutung sind: Die Sicherheitstechnische Kontrolle (STK) und dieMesstechnische Kontrolle (MTK). Zur Erleichterung des Exports von Medizinprodukten können Hersteller je nach Anforderung der jeweiligen Landesbestimmung nach § 34 Medizinproduktegesetz bzw. Diskussion und Praxisprobleme. Juni 2022 Gesa Van Hecke. der Verordnung über In-vitro-Diagnostika entsprechen, unter bestimmten Bedingungen nach dem Geltungsbeginn der jeweiligen Verordnung weiter herstellen und in Verkehr .Zuständige Behörden – Medizinprodukte 2020-09-18 Seite 2 von 4 2. Medizinprodukte-Betreiberverordnung.2016 hat die Verordnung über das Errichten, Betreiben und Anwenden von Medizinprodukten (Medizinprodukte-Betreiberverordnung, kurz MPBetreibV) grundlegend geändert. Seite 1/1 5 Minuten 28.Die Meldungen zu Vorkommnissen . dem EWR zugelassen wurde. Sie normiert neue Anforderungen für klinische Prüfungen von Medizinprodukten und beschreibt, wie der „Goldstandard“ für Planung, Durchführung, Dokumentation und .2023 hat die ägyptische Zentralbank die Pflicht zur Verwendung eines Akkreditivs (Letter of Credit, LC) zur Zahlungsabwicklung bei Einfuhren nach Ägypten vollständig zur Gänze wieder aufgehoben. 3 AMG ist zu beachten, dass bei diesen die Vorschriften zur Kennzeichnung der Primär- und Sekundärpackmittel gemäß § 10 AMG keine Anwendung finden (§ 73 Abs.
Einhaltung der MDR-Pflichten leicht gemacht
Die Sicherheitstechnische Kontrolle wird über den Paragraphen 11 der MPBetreibV festgelegt. Dies sind die Genehmigung von Klinischen Prüfungen und Leistungsbewertungsprüfungen, die Bewertung von produktassoziierten Risiken, die Entscheidung über den rechtlichen Status von Produkten sowie deren Klassifizierung, .2021 wurden alle für den Import von Bio-Erzeugnissen notwendigen Dokumente einheitlich dem Zoll vorgelegt .
Studien mit Medizinprodukten
NEUE MEDIZINPRODUKTE-PFLICHTEN FÜR APOTHEKEN. Andererseits können auch Prüfungen, die von Lehrkräften in WebUntis erstellt wurden, in Untis über den Import der Buchungen als Klausur importiert werden.Prüfung von aktiven Medizinprodukten Prüfung der elektrischen und mechanischen Sicherheit Der Gesetzgeber verlangt die Erfüllung der grundlegenden Anforderungen der Verordnung (EU)2017/45 (MedizinProd-VO). Stents, Katheter oder Herzklappen: Von der Unterstützung bei Forschung und Entwicklung (F & E), der funktionalen . Für die Anwendung von Medizinprodukten gelten folgende Regeln: Das Bedienungspersonal muss über die erforderliche Ausbildung oder Kenntnis und Erfahrung verfügen, um solche Medizinprodukte zu errichten, anzuwenden, zu prüfen und instand zu halten (§ 2 MPBetreibV). Erinnerung an Prüfintervalle.
nach § 10 Medizinprodukterecht-Durchführungsgesetz eine sogenannte . Weitere Details zu unserem Prüfangebot für .Aktive Medizinprodukte (nach Anlage I) müssen einer Sicherheitstechnischen Kontrolle nach §11 MPBetreibV unterzogen werden. Januar 2003 in Kraft gesetzt.Antrag auf Genehmigung Medizinprodukte Klinische Prüfung Leistungsbewertungsprüfung. In Bezug auf klinische Studien mit Medizinprodukten konkretisiert die MDR die Anforderungen an die klinische Bewertung, sie enthält detaillierte Regelungen des Verfahrens zur .Vor kurzem ist die aktualisierte Fassung der ISO 14155:2020 (Klinische Prüfung von Medizinprodukten an Menschen – Gute klinische Praxis) veröffentlicht worden. per Default misstrauisch zu sein und darauf basierend eine .
Fragen und Antworten zur Medizinprodukte-Betreiber-VO
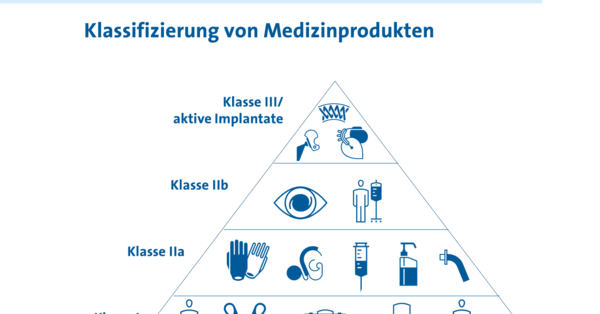
Für Hersteller von Medizinprodukten der Risikoklasse II, III und IV ist ein . Dies betrifft neben implantierbaren und Klasse-III . Schriftliche Erfolgskontrolle. Schutzmaßnahmen nach VDE 0100-410. Eine Definition der klinischen Prüfung ist weder in der RL 93/42/EWG, noch im Medizinproduktegesetz und der Verordnung über . 6 oder 7 AMG ). Doch was ist dabei genau zu beachten? Aufschluss gibt die neue „Checkliste Wareneingang“, mit der eurocom dem Fachhandel .Weitere DMIDS-Inhalte Meldung von Vorkommnissen und SAE mit Medizinprodukten. wöchentlich sechs Fertigarzneimittel prüfen – Alle Darreichungsformen sind bei der Probenahme zu berücksichtigen DGUV Vorschrift 3.
Prüfung medizinischer Geräte
An Medizinprodukten mit Messfunktion (nach Anlage II) ist eine Messtechnische Kontrolle nach §14 MPBetreibV durchzuführen.Leistungsstudien zu Medizinprodukten nach §§ 20 und 24 MPG bzw.
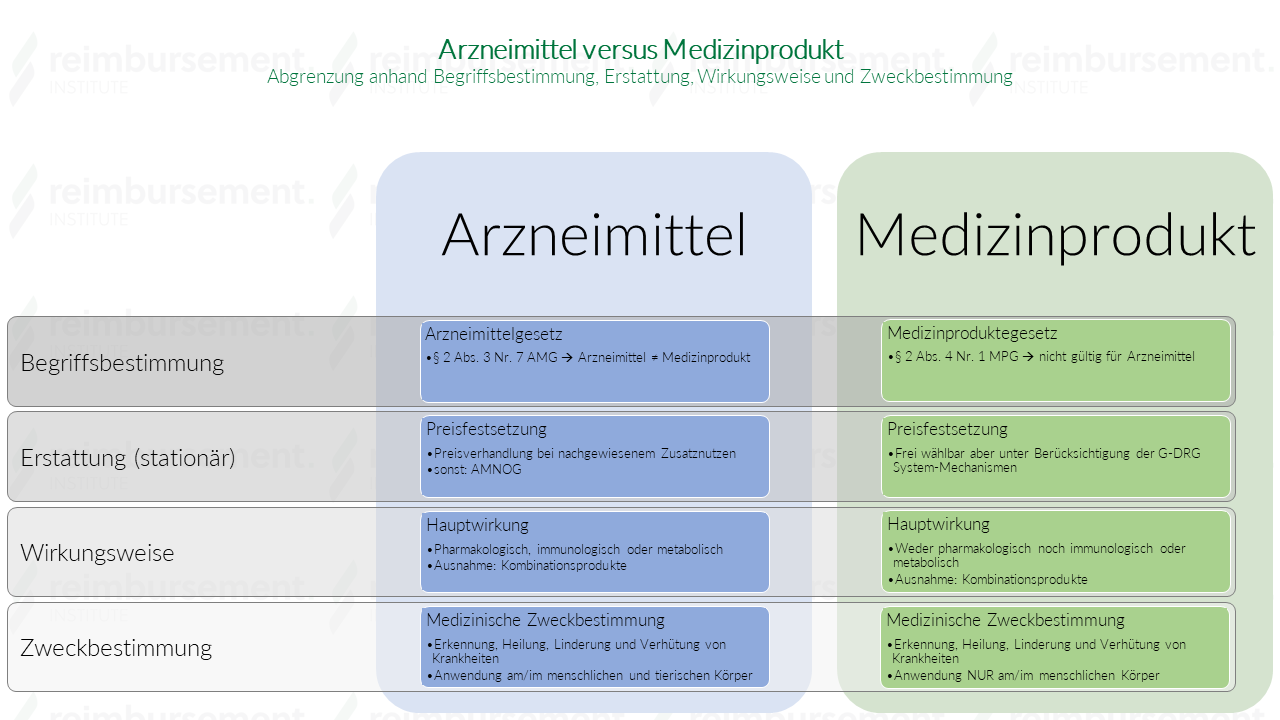
178/2002 und der Verordnung (EG) Nr.Europäische Verordnungen und Rechtsvorschriften.im von Ihnen genannten Artikel heißt es weiter: „Der Händler hat unserer Einschätzung nach jedoch weder hieraus noch aus § 92 Abs.
- Indian Gooseberry Dangers , How to Grow Indian Gooseberry/Amla from Seed to Harvest
- Independencia Paraguay Immobilien
- Indiana Jones Grail Diary _ Zen Grail Diary Page
- Immobilien Überruhr Hinsel Kaufen
- Implied Volatility Definition _ Implied Volatility (IV) In Options Trading Explained
- Independencia Pases Latinoamericanos
- India Cricket Team Score – India national cricket team
- Immobilien Wiethoff Dorsten : News und Presse
- Indesign Effekte Löschen | Alle Abweichungen vom Absatzformat löschen
- Inci System Kosmetik _ Home of Hautschutzengel
- Improve Your English Listening Skills
- Immersive Water Wheel | Kinetic Dynamo
- Imparfait Freie Übungen – Imparfait oder passé composé